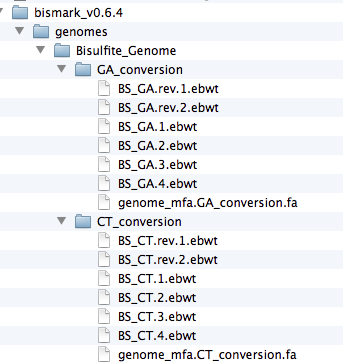
(I) Running bismark_genome_preparation USAGE:bismark_genome_preparation [options] <path_to_genome_folder>
A typical genome indexing could look like this:
/bismark/bismark_genome_preparation --path_to_bowtie /usr/local/bowtie/ -- verbose /data/genomes/homo_sapiens/GRCh37/
COMMAND
./bismark_genome_preparation --path_to_bowtie /Volumes/Bay3/Software/bowtie/ --verbose ./genomes
#note only single fasta placed in genomes folder.
 --
--
# created these files
# next step
(II) Running bismark
USAGE: bismark [options] <genome_folder> {-1 <mates1> -2 <mates2> |
<singles>}
Typical alignment example (tolerating one non-bisulfite mismatch per read):
bismark -n 1 -l 50 /data/genomes/homo_sapiens/GRCh37/ test_dataset.fastq
February 06, 2012
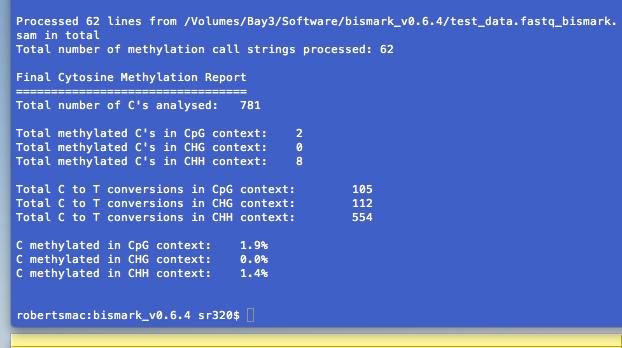
This will produce two output files:
(a) test_dataset.fastq_bismark.sam (contains all alignments plus methylation call strings)
(b) test_dataset.fastq_bismark_mapping_report.txt (contains alignment and methylation summary)
NOTE: In order to work properly the current working directory must contain the sequence files to be analysed.
COMMAND
./bismark -n 1 -l 50 ./genomes/ /Volumes/Bay3/Software/bismark_v0.6.4/filtered_P_Ab_CO2_ACTTGA_L001_R1_trimmed.fastq
#
#confirmed duplicate IDs, using Galaxy to resolve.
RESOLVED
NOW HAVE TO START OVER!!!!!!
VIDEO OF REDO
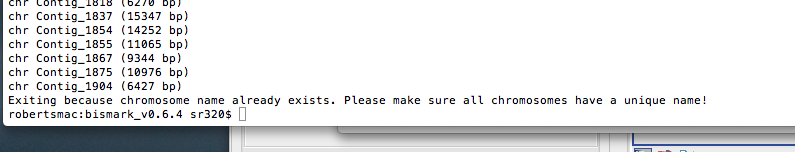
Failed as this error after bimark run
---
chr 28523ConsensusfromContig72814 (5751 bp)
chr 28524ConsensusfromContig72947 (6122 bp)
chr 28525ConsensusfromContig73208 (5020 bp)
chr 28526ConsensusfromContig73216 (6167 bp)
Reading in the sequence file /Volumes/Bay3/Software/bismark_v0.6.4/filtered_Unlabeled_NoIndex_L003_R1_trimmed.fastq
The same seq ID occurred more than twice in a row
Trouble shooting
trying bsSeqref
--
Trouble shooting
running test data
#win
appears to be problem with reads
going to try to give unique IDs
re importing and sending to Galaxy
00
re import should be faster
-next day
now need to trim and export
Exported to Scratch Drive, now ready to try Bismark again